Tutorial EPH Temperature-Dependence (Legacy)¶
Electron-phonon Temperature-DEPendence of the Electronic Structure.¶
This tutorial aims at showing how to get the following physical properties, for periodic solids:
-
The zero-point-motion renormalization (ZPR) of eigenenergies
-
The temperature-dependence of eigenenergies
-
The lifetime/broadening of eigenenergies
It should take about 1 hour.
WARNING : This tutorial concerns an old procedure to obtain the temperature-dependence of the electronic structure, that is why it is labelled “legacy”.
For the theory related to the temperature-dependent calculations, please read the following papers: [Ponce2015], [Ponce2014] and [Ponce2014a].
There are three ways to compute the temperature dependence with Abinit:
-
Using Anaddb: historically the first implementation.
-
Using post-processing python scripts: This way provides more options and is more efficient (less disk space, less memory demanding). This option requires Netcdf (both in Abinit and python). In this tutorial, we only focus on this approach.
-
Using an interpolation of the perturbed potential: This new way is covered in the ZPR and T-dependent band structures tutorial.
Important
In order to run the python script you need:
- python 2.7.6 or higher, python3 is not supported
- numpy 1.7.1 or higher
- netCDF4 and netCDF4 for python
- scipy 0.12.0 or higher
This can be done with:
sudo apt-get install netcdf-bin
sudo apt-get install python-dev
pip install numpy
pip install scipy
pip install netcdf4
A list of configuration files for clusters is available in the abiconfig repository
If you have a prebuilt abinit executable, use:
./abinit -b
to get the list of libraries/options activated in the build.
You should see netcdf in the TRIO flavor section:
=== Connectors / Fallbacks ===
LINALG flavor : netlib
FFT flavor : goedecker
HDF5 : yes
NetCDF : yes
NetCDF Fortran : yes
LibXC : yes
Wannier90 : no
Note
Supposing you made your own installation of ABINIT, the input files to run the examples are in the ~abinit/tests/ directory where ~abinit is the absolute path of the abinit top-level directory. If you have NOT made your own install, ask your system administrator where to find the package, especially the executable and test files.
In case you work on your own PC or workstation, to make things easier, we suggest you define some handy environment variables by executing the following lines in the terminal:
export ABI_HOME=Replace_with_absolute_path_to_abinit_top_level_dir # Change this line
export PATH=$ABI_HOME/src/98_main/:$PATH # Do not change this line: path to executable
export ABI_TESTS=$ABI_HOME/tests/ # Do not change this line: path to tests dir
export ABI_PSPDIR=$ABI_TESTS/Psps_for_tests/ # Do not change this line: path to pseudos dir
Examples in this tutorial use these shell variables: copy and paste
the code snippets into the terminal (remember to set ABI_HOME first!) or, alternatively,
source the set_abienv.sh script located in the ~abinit directory:
source ~abinit/set_abienv.sh
The ‘export PATH’ line adds the directory containing the executables to your PATH so that you can invoke the code by simply typing abinit in the terminal instead of providing the absolute path.
To execute the tutorials, create a working directory (Work*) and
copy there the input files of the lesson.
Most of the tutorials do not rely on parallelism (except specific tutorials on parallelism). However you can run most of the tutorial examples in parallel with MPI, see the topic on parallelism.
Visualisation tools are NOT covered in this tutorial. Powerful visualisation procedures have been developed in the Abipy context, relying on matplotlib. See the README of Abipy and the Abipy tutorials.
1 Calculation of the ZPR of eigenenergies at q=Γ.¶
The reference input files for this tutorial are located in
~abinit/tests/tutorespfn/Input and the corresponding reference output files
are in ~abinit/tests/tutorespfn/Refs.
The prefix for files is teph_tdep_legacy. As usual, we use the shorthand ~abinit to indicate
the root directory where the abinit package has been deployed, but most often
consider the paths relative to this directory.
First, examine the tests/tutorespfn/Input/teph_tdep_legacy_1.abi input file.
# C in diamond structure. ndtset 3 elph2_imagden 0.1 eV # Imaginary shift of the denominator of the sum-over-states # in the perturbation denominator. Usual value is 0.1 eV to reproduce # experimental broadening at 300K. Increasing the value help the # convergence with respect to the number of q-points. ngkpt 2 2 2 # Underconverged : k-grid should be at least 4x4x4 for diamond to be converged. nshiftk 1 shiftk 0.0 0.0 0.0 # Ground state density tolvrs1 1.0d-8 # Underconverged : tolvrs 1.0d-18 should be used for converged results # Non self-consistent calculation with an arbitrary q point (here Gamma) getden2 1 iscf2 -2 nqpt2 1 qpt2 0.0 0.0 0.0 nbdbuf2 2 tolwfr2 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results # Computation at Gamma getwfk3 1 getwfq3 2 nqpt3 1 qpt3 0.0 0.0 0.0 # Calculation at Gamma ieig2rf3 5 # Static eigenvalues corrections using DFPT (Sternheimer) smdelta3 1 # Flag required to produce the _EIGI2D used to # compute the lifetime of electronic levels. # smdelta = 1 ==> Fermi-Dirac smearing. nbdbuf3 2 # 2 buffer bands. RF converges much faster. rfphon3 1 # Do phonon response tolwfr3 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results # Cell dependent parameters acell 3*6.675 rprim 0 .5 .5 .5 0 .5 .5 .5 0 nsym 1 # Disable symmetries. K-point symmetries are not yet working. kptopt 3 # Disable TR symmetry natom 2 typat 1 1 xred 3*0.0 3*0.25 nband 12 ntypat 1 znucl 6 diemac 6 ecut 37 enunit 2 istwfk *1 pp_dirpath "$ABI_PSPDIR" pseudos "Pseudodojo_nc_sr_04_pw_standard_psp8/C.psp8" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% teph_tdep_legacy_1.abo, tolnlines= 20, tolabs= 5.000e-05, tolrel= 5.000e-04, fld_options=-medium #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = S. Ponc\'e #%% keywords = NC, DFPT, EPH_OLD #%% description = Temperature dependence calculation of diamond. #%%<END TEST_INFO>
Note that there are three datasets (ndtset=3). The first dataset corresponds to a standard self-consistent calculation, with an unshifted eight k-point grid, producing e.g. the ground-state eigenvalue file teph_tdep_legacy_1o_DS1_EIG.nc , as well as the density file teph_tdep_legacy_1o_DS1_DEN. The latter is read (getden2=1) to initiate the second dataset calculation, which is a non-self-consistent run, specifically at the Gamma point only (there is no real recomputation with respect to the dataset 1, it only extract a subset of the eight k-point grid). This second dataset produces the wavefunction file teph_tdep_legacy_1o_DS2_WFQ, that is read by the third dataset (getwfq3=2), as well as the teph_tdep_legacy_1o_DS1_WFK file from the first dataset (getwfk3=1).
The third dataset corresponds to a DFPT phonon calculation (rfphon3=1) with displacement of all atoms (rfatpol3= 1 2) in all directions (rfdir3= 1 1 1), which are the default values. This induces the creation of the Derivative DataBase file teph_tdep_legacy_1o_DS3_DDB. The electron-phonon matrix elements are produced because of ieig2rf3=5 , this option generating the needed netCDF files teph_tdep_legacy_1o_DS3_EIGR2D.nc and teph_tdep_legacy_1o_DS3_GKK.nc .
In order to run abinit, we suggest that you create a working directory, why not call it Work,
as subdirectory of ~abinit/tests/tutorespfn/Input, then
copy/modify the relevant files. Explicitly:
cd ~abinit/tests/tutorespfn/Input
mkdir Work
cd Work
cp ../teph_tdep_legacy*in .
Finally, issue
abinit teph_tdep_legacy_1.abi
The calculation will produce several _EIG.nc, _DDB, EIGR2D.nc and EIGI2D.nc files, that contain respectively the eigenvalues (GS or perturbed), the second-order derivative of the total energy with respect to two atomic displacements, the electron-phonon matrix elements used to compute the renormalization of the eigenenergies and the electron-phonon matrix elements used to compute the lifetime of the electronic states.
You can now copy three post-processing python files from ~abinit/scripts/post_processing/temperature-dependence . Make sure you are in the directory containing the output files produced by the code and issue:
cp ~abinit/scripts/post_processing/temperature-dependence/temperature_final.py .
cp ~abinit/scripts/post_processing/temperature-dependence/rf_final.py .
cp ~abinit/scripts/post_processing/plot_bs.py .
in which ~abinit has been replaced by the proper path.
You can then simply run the python script with the following command:
python temperature_final.py
and enter the information asked by the script, typically the following (data contained in ~abinit/tests/tutorespfn/Input/teph_tdep_legacy_1_temperature.in):
1 # Number of cpus
2 # Static ZPR computed in the Allen-Heine-Cardona theory
temperature_1 # Prefix for output files
0.1 # Value of the smearing parameter for AHC (in eV)
0.1 # Gaussian broadening for the Eliashberg function and PDOS (in eV)
0 0.5 # Energy range for the PDOS and Eliashberg calculations (in eV)
0 1000 50 # min, max temperature and temperature step
1 # Number of Q-points we have (here we only computed $\Gamma$)
teph_tdep_legacy_1o_DS3_DDB # Name of the response-funtion (RF) DDB file
teph_tdep_legacy_1o_DS2_EIG.nc # Eigenvalues at $\mathbf{k+q}$
teph_tdep_legacy_1o_DS3_EIGR2D.nc # Second-order electron-phonon matrix element
teph_tdep_legacy_1o_DS3_GKK.nc # Name of the 0 GKK file
teph_tdep_legacy_1o_DS1_EIG.nc # Name of the unperturbed EIG.nc file with Eigenvalues at $k$
Alternatively, copy this example file in the Work directory if not yet done, and then run
python temperature_final.py < teph_tdep_legacy_1_temperature.in
1 2 temperature_1 0.1 0.1 0 0.5 0 1000 50 1 teph_tdep_legacy_1o_DS3_DDB teph_tdep_legacy_1o_DS2_EIG.nc teph_tdep_legacy_1o_DS3_EIGR2D.nc teph_tdep_legacy_1o_DS3_GKK.nc teph_tdep_legacy_1o_DS1_EIG.nc
Warning
Remember to install the libraries required by the script before running.
For pip, use:
pip install netcdf4
or:
conda install netcdf4
if you are using conda
You should see on the screen an output similar to:
Start on 21/12/2020 at 15h21
____ ____ _ _
| _ \| _ \ | |_ ___ _ __ ___ _ __ ___ _ __ __ _| |_ _ _ _ __ ___
| |_) | |_) |____| __/ _ \ '_ ` _ \| '_ \ / _ \ '__/ _` | __| | | | '__/ _ \
| __/| __/_____| || __/ | | | | | |_) | __/ | | (_| | |_| |_| | | | __/
|_| |_| \__\___|_| |_| |_| .__/ \___|_| \__,_|\__|\__,_|_| \___|
|_| Version 1.5
This script compute the static/dynamic zero-point motion
and the temperature dependence of eigenenergies due to electron-phonon interaction.
The electronic lifetime can also be computed.
WARNING: The first Q-point MUST be the Gamma point.
Enter the number of cpu on which you want to multi-thread
Define the type of calculation you want to perform. Type:
1 if you want to run a non-adiabatic AHC calculation
2 if you want to run a static AHC calculation
3 if you want to run a static AHC calculation without control on active space (not recommended !)
Note that for 1 & 2 you need _EIGR2D.nc and _GKK.nc files obtained through ABINIT option "ieig2rf 5"
Enter name of the output file
Enter value of the smearing parameter for AHC (in eV)
Enter value of the Gaussian broadening for the Eliashberg function and PDOS (in eV)
Enter the energy range for the PDOS and Eliashberg calculations (in eV): [e.g. 0 0.5]
Introduce the min temperature, the max temperature and the temperature steps. e.g. 0 200 50 for (0,50,100,150)
Enter the number of Q-points you have
Enter the name of the 0 DDB file
Enter the name of the 0 eigq file
Enter the name of the 0 EIGR2D file
Enter the name of the 0 GKK file
Enter the name of the unperturbed EIG.nc file at Gamma
Inside the dynamic_zpm_temp def
Q-point: 0 with wtq = 1.0 and reduced coord. [0. 0. 0.]
WARNING: An eigenvalue is negative with value: -2.8630004909173537e-10 ... but proceed with value 0.0
Now compute active space ...
Now compute generalized g2F Eliashberg electron-phonon spectral function ...
End on 21/12/2020 at 15 h 21
Runtime: 0 seconds (or 0.0 minutes)
The python code has generated the following files:
- temperature_1.txt
- This text file contains the zero-point motion renormalization (ZPR) at each k-point for each band. It also contain the evolution of each band with temperature at k=\Gamma. At the end of the file, the Fan/DDW contribution is also reported.
- temperature_1_EP.nc
- This netcdf file contains a number for each k-point, for each band and each temperature. The real part of this number is the ZPR correction and the imaginary part is the lifetime.
We can for example visualize the temperature dependence at k=\Gamma of the LUMO bands
(Band: 4 section in the temperature_1.txt file, that you can examine)
with the contribution of only q=\Gamma.
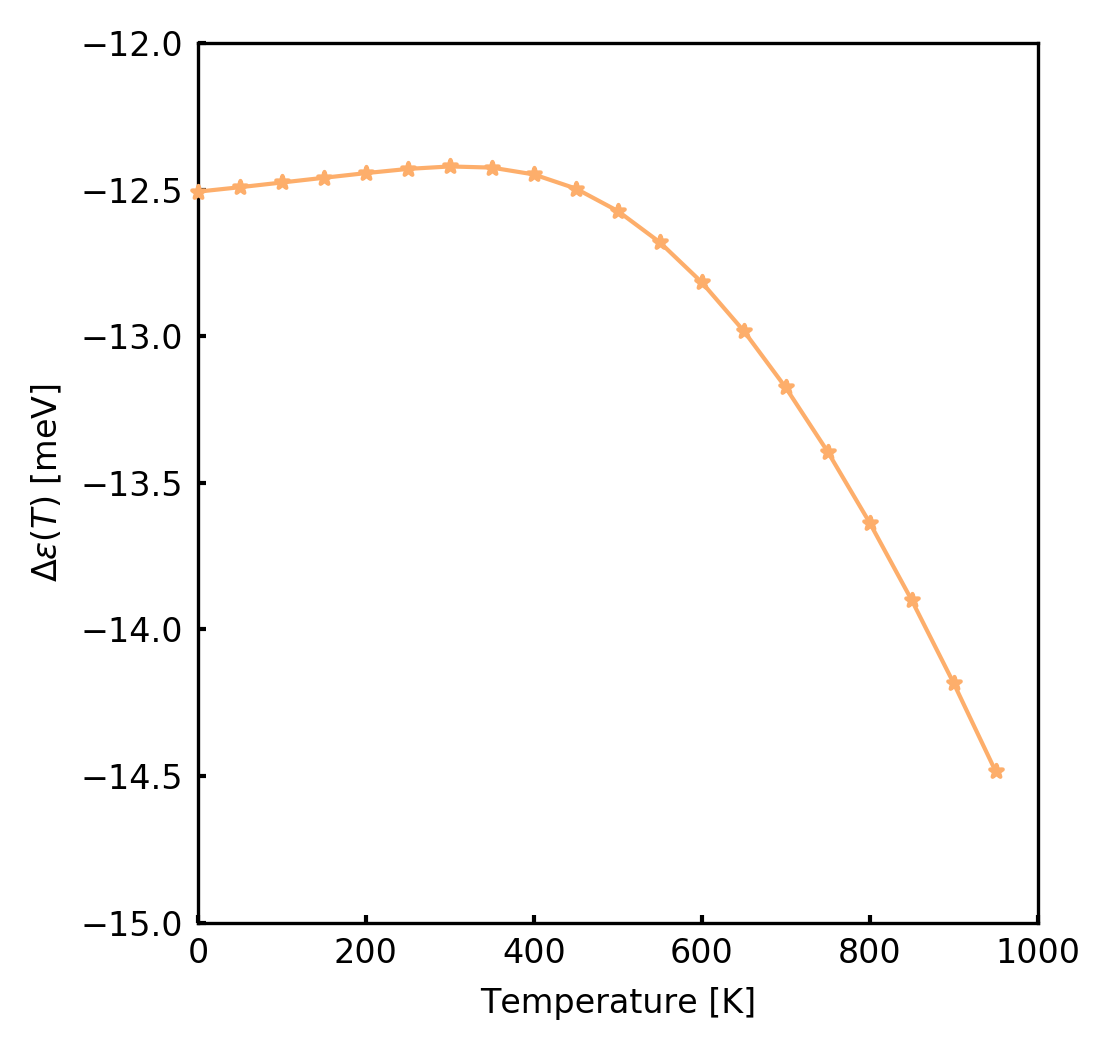
As you can see, the LUMO correction goes down with temperature. If the calculations were converged, the HOMO eigenenergies correction should go up with temperature. In general, the ZPR correction as well as their temperature dependence usually closes the gap of semiconductors.
As usual, checking whether the input parameters give converged values is of course important.
2 Converging the calculation with respect to the grid of phonon wavevectors¶
Convergence studies with respect to most of the parameters will rely on obvious modifications of the input file detailed in the previous section. However, using more than one q-point phonon wavevector needs a non-trivial generalisation of this procedure. This is because each q-point needs to be treated in a different dataset in the current version of ABINIT.
The code can perform the q-wavevector integration either with random q-points or homogenous Monkhorst-Pack meshes. Both grids have been used in the Ref. [Ponce2014], see e.g. Fig. 3 of this paper.
For the random integration method you should create a script that generates random q-points, perform the Abinit calculations at these points, gather the results and analyze them. The temperature_final.py script will detect that you used random integration thanks to the weight of the q-point stored in the _EIGR2D.nc file and perform the integration accordingly. The random integration converges slowly but in a smooth manner.
However, since this method is a little bit less user-friendly than the one based on homogeneous grids, we will focus on this homogenous integration. In this case, the user must specify in the ABINIT input file the homogeneous q-point grid, using input variables like ngqpt, qptopt, shiftq, nshiftq, …, i.e. variables whose names are similar to those used to specify the k-point grid (for electrons).
There are several difficulties here. First, since we focus on the k=\Gamma point, we expect to be able to use symmetries to decrease the computational load, as \Gamma is invariant under all symmetry operations of the crystal. The symmetry operations of the crystal will be used to decrease the number of q-wavevectors, but they cannot be used as well to decrease the k-point grid during the corresponding self-consistent phonon computation. How this different behaviour of k-grids and q-grids can be handled by ABINIT ? By convention, in such case, with nsym=1 the k-point grid will be generated in the Full Brillouin zone, without use of symmetries, while the q-point grid with qptopt=1 with be generated in the irreducible Brillouin Zone, despite nsym=1. In order to generate q-point grids that are not folded in the irreducible Brillouin Zone, one need to use another value of qptopt. In particular qptopt=3 has to be used to generate q points in the full Brillouin zone.
Second, the number of ABINIT datasets is expected to be given in the input file, by the user, but not determined on-the-flight by ABINIT. Still, this number of datasets is determined by the number of q points. Thus, the user will have to compute it before being able to launch the real q-point calculations, since it determines ndtset.
How to determine the number of irreducible q points ?
Well, the easiest procedure is to compute it for an equivalent k-point grid, by a quick run.
An example will clarify this. Suppose that one is looking for the number of q-points corresponding to
ngqpt 4 4 4
qptopt 1
nshiftq 1
shiftq 0.0 0.0 0.0
One make a quick ABINIT run with tests/tutorespfn/Input/teph_tdep_legacy_2.abi. Note that several input variables have been changed with respect to tests/tutorespfn/Input/teph_tdep_legacy_1.abi:
ndtset 1
nstep 0
prtebands 0
ngkpt 4 4 4
nshiftk 1
shiftk 0.0 0.0 0.0
nsym 0
In this example, the new values of ndtset and nstep, and the definition of prtebands allow a fast run (nline==0 might be specified as well, or even, the run might be interrupted after a few seconds, since the number of k points is very quickly available). Then, the k-point grid is specified thanks to ngkpt, nshiftk, shiftk, replacing the corresponding input variables for the q-point grid. The use of symmetries has been reenabled thanks to nsym=0.
To run it, issue:
abinit teph_tdep_legacy_2.abi
Now, the number of points can be seen in the output file :
nkpt 8
the list of these eight k-points being given in
kpt 0.00000000E+00 0.00000000E+00 0.00000000E+00
2.50000000E-01 0.00000000E+00 0.00000000E+00
5.00000000E-01 0.00000000E+00 0.00000000E+00
2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 2.50000000E-01
We are now ready to launch the determination of the _EIG.nc, _DDB, EIGR2D.nc and EIGI2D.nc files, with 8 q-points. As for the \Gamma calculation of the previous section, we will rely on three datasets for each q-point. This permits a well-structured set of calculations, although there is some redundancy. Indeed, the first of these datasets will correspond to an unperturbed ground-state calculation identical for all q. It is done very quickly because the converged wavefunctions are already available. The second dataset will correspond to a non-self-consistent ground-state calculation at k+q (it is also quick thanks to previously available wavefunctions), and the third dataset will correspond to the DFPT calculations at k+q (this is the CPU intensive part) .
So, compared to the first run in this tutorial, we have to replace
ndtset 3 by ndtset 24 udtset 8 3
in the input file tests/tutorespfn/Input/teph_tdep_legacy_3.abi, and adjusted accordingly all input variables that were dataset-dependent.
# C in diamond structure. ndtset 24 udtset 8 3 iqpt:? 1 # Index of the first q-point of this file (useful if you split your # input files elph2_imagden 0.1 eV # Imaginary shift of the denominator of the sum-over-states # in the perturbation denominator. Usual value is 0.1 eV to reproduce # experimental broadening at 300K. Increasing the value help the # convergence with respect to the number of q-points. ngkpt 2 2 2 # Underconverged : k-grid should be at least 4x4x4 for diamond to be converged. nshiftk 1 shiftk 0.0 0.0 0.0 ngqpt 4 4 4 # Should be converged upon qptopt 1 nshiftq 1 shiftq 0.0 0.0 0.0 # Ground state density iqpt+? 1 tolvrs?1 1.0d-8 # Underconverged : tolvrs 1.0d-18 should be used for converged results # Non self-consistent calculation with an arbitrary q point (here Gamma) getden?2 -1 iscf?2 -2 nqpt?2 1 nbdbuf?2 2 tolwfr?2 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results kptopt?2 3 # Computation at Gamma getwfk?3 -2 getwfq?3 -1 nqpt?3 1 ieig2rf?3 5 # Static eigenvalues corrections using DFPT (Sternheimer) smdelta?3 1 # Flag required to produce the _EIGI2D used to # compute the lifetime of electronic levels. # smdelta = 1 ==> Fermi-Dirac smearing. nbdbuf?3 2 # 2 buffer bands. RF converges much faster. rfphon?3 1 # Do phonon response tolwfr?3 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results kptopt?3 3 # Cell dependent parameters acell 3*6.675 rprim 0 .5 .5 .5 0 .5 .5 .5 0 nsym 1 # Disable symmetries. Symmetries are not yet in production. natom 2 typat 1 1 xred 3*0.0 3*0.25 nband 12 ntypat 1 znucl 6 diemac 6 ecut 37 enunit 2 istwfk *1 pp_dirpath "$ABI_PSPDIR" pseudos "Pseudodojo_nc_sr_04_pw_standard_psp8/C.psp8" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% teph_tdep_legacy_3.abo, tolnlines= 30, tolabs= 5.000e-05, tolrel= 5.000e-04, fld_options=-medium #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = S. Ponc\'e #%% keywords = NC, DFPT, EPH_OLD #%% description = Temperature dependence calculation of diamond. #%%<END TEST_INFO>
Please, refer to the explanation of the usage of a double-loop of datasets if you are confused about the meaning of udtset, and the usage of the corresponding metacharacters. We have indeed also introduced
iqpt:? 1
iqpt+? 1
that translates into
iqpt11 1
iqpt12 1
iqpt13 1
iqpt21 2
iqpt22 2
iqpt23 2
iqpt31 3
...
allowing to perform calculations for three datasets at each q-point.
Then issue:
abinit teph_tdep_legacy_3.abi
This is a significantly longer ABINIT run (still less than two minutes), also producing many files.
When the run is finished, copy the file tests/tutorespfn/Input/teph_tdep_legacy_3_temperature.in in the working directory (if not yet done) and launch the python script with:
./temperature_final.py < teph_tdep_legacy_3_temperature.in
1 2 temperature_3 0.1 0.1 0 0.5 0 1000 50 8 teph_tdep_legacy_3o_DS13_DDB teph_tdep_legacy_3o_DS23_DDB teph_tdep_legacy_3o_DS33_DDB teph_tdep_legacy_3o_DS43_DDB teph_tdep_legacy_3o_DS53_DDB teph_tdep_legacy_3o_DS63_DDB teph_tdep_legacy_3o_DS73_DDB teph_tdep_legacy_3o_DS83_DDB teph_tdep_legacy_3o_DS12_EIG.nc teph_tdep_legacy_3o_DS22_EIG.nc teph_tdep_legacy_3o_DS32_EIG.nc teph_tdep_legacy_3o_DS42_EIG.nc teph_tdep_legacy_3o_DS52_EIG.nc teph_tdep_legacy_3o_DS62_EIG.nc teph_tdep_legacy_3o_DS72_EIG.nc teph_tdep_legacy_3o_DS82_EIG.nc teph_tdep_legacy_3o_DS13_EIGR2D.nc teph_tdep_legacy_3o_DS23_EIGR2D.nc teph_tdep_legacy_3o_DS33_EIGR2D.nc teph_tdep_legacy_3o_DS43_EIGR2D.nc teph_tdep_legacy_3o_DS53_EIGR2D.nc teph_tdep_legacy_3o_DS63_EIGR2D.nc teph_tdep_legacy_3o_DS73_EIGR2D.nc teph_tdep_legacy_3o_DS83_EIGR2D.nc teph_tdep_legacy_3o_DS13_GKK.nc teph_tdep_legacy_3o_DS23_GKK.nc teph_tdep_legacy_3o_DS33_GKK.nc teph_tdep_legacy_3o_DS43_GKK.nc teph_tdep_legacy_3o_DS53_GKK.nc teph_tdep_legacy_3o_DS63_GKK.nc teph_tdep_legacy_3o_DS73_GKK.nc teph_tdep_legacy_3o_DS83_GKK.nc teph_tdep_legacy_3o_DS11_EIG.nc
Examination of the same HOMO and LUMO bands at k=\Gamma for a 4x4x4 q-point grid gives a very different result than previously. The zero-point renormalization (ZPR) is the change of the bandgap at 0 K and was (band 5 - band 4):
-0.012507 - 0.017727 = -0.030234 eV
and is now:
-0.351528 - 0.095900 = -0.447428 eV
This means that the bandgap was closing by 30 meV at 0 K and is now closing by 447 meV at 0 K. For comparison, the converged direct bandgap ZPR of diamond is 438.6 meV from Ref. [Ponce2015].
As a matter of fact, diamond requires an extremely dense q-point grid (40x40x40) to be converged. On the bright side, each q-point calculation is independent and thus the parallel scaling is ideal. Running separate jobs for different q-points is quite easy thanks to the dtset approach.
3 Calculation of the eigenenergy corrections along high-symmetry lines¶
The calculation of the electronic eigenvalue correction due to electron-phonon coupling along high-symmetry lines requires the use of 6 datasets per q-point. Moreover, the choice of an arbitrary k-wavevector breaks all symmetries of the crystal. Different datasets are required to compute the following quantites:
\Psi^{(0)}_{kHom} : The ground-state wavefunctions on the Homogeneous k-point sampling.
\Psi^{(0)}_{kBS} : The ground-state wavefunctions computed along the bandstructure k-point sampling.
\Psi^{(0)}_{kHom+q} : The ground-state wavefunctions on the shifted Homogeneous k+q-point sampling.
n^{(1)} : The perturbed density integrated over the homogeneous k+q grid.
\Psi^{(0)}_{kBS+q} : The ground-state wavefunctions obtained from reading the perturbed density of the previous dataset.
Reading the previous quantity we obtain the el-ph matrix elements along the bandstructure with all physical quantities integrated over a homogeneous grid.
We will use the tests/tutorespfn/Input/teph_tdep_legacy_4.abi input file
# C in diamond structure. ndtset 48 udtset 8 6 iqpt:? 1 # Index of the first q-point of this file (useful if you split your # input files elph2_imagden 0.1 eV # Imaginary shift of the denominator of the sum-over-states # in the perturbation denominator. Usual value is 0.1 eV to reproduce # experimental broadening at 300K. Increasing the value help the # convergence with respect to the number of q-points. ngkpt 2 2 2 # Underconverged : k-grid should be at least 4x4x4 for diamond to be converged. nshiftk 1 shiftk 0.0 0.0 0.0 ngqpt 2 2 2 # Should be converged upon qptopt 3 nshiftq 1 shiftq 0.0 0.0 0.0 # Ground state density iqpt+? 1 tolvrs?1 1.0d-8 # Underconverged : tolvrs 1.0d-18 should be used for converged results # Non self-consistent calculation following high sym k path getden?2 -1 iscf?2 -2 getwfk?2 -1 nstep?2 50 tolwfr?2 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results kptopt?2 -9 ndivsm?2 5 kptbounds?2 1/2 0.0 0.0 # L 0.0 0.0 0.0 # Gamma 0.0 1/2 1/2 # X 1/4 1/2 3/4 # W 3/8 3/8 3/4 # K 1/2 1/2 1/2 # L 1/4 1/2 3/4 # W 1/2 1/2 1.0 # X 3/8 3/8 3/4 # K 0.0 0.0 0.0 # Gamma # Non self-consistent calculation with an arbitrary q point getden?3 -2 getwfk?3 -2 iscf?3 -2 nqpt?3 1 nbdbuf?3 2 tolwfr?3 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results kptopt?3 1 # Computation at q getwfk?4 -3 getwfq?4 -1 nqpt?4 1 ieig2rf?4 5 # Static eigenvalues corrections using DFPT (Sternheimer) smdelta?4 1 # Flag required to produce the _EIGI2D used to # compute the lifetime of electronic levels. # smdelta = 1 ==> Fermi-Dirac smearing. nbdbuf?4 2 # 2 buffer bands. RF converges much faster. rfphon?4 1 # Do phonon response tolwfr?4 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results kptopt?4 3 # Non self-consistent calculation following high sym k path nqpt?5 1 getden?5 -4 iscf?5 -2 get1den?5 -1 getwfk?5 -3 nbdbuf?5 2 # 2 buffer bands. RF converges much faster. nstep?5 1 # We do not want mixing (self-consistent) tolwfr?5 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results kptopt?5 -9 ndivsm?5 5 kptbounds?5 1/2 0.0 0.0 # L 0.0 0.0 0.0 # Gamma 0.0 1/2 1/2 # X 1/4 1/2 3/4 # W 3/8 3/8 3/4 # K 1/2 1/2 1/2 # L 1/4 1/2 3/4 # W 1/2 1/2 1.0 # X 3/8 3/8 3/4 # K 0.0 0.0 0.0 # Gamma # Computation at an other q point nqpt?6 1 getden?6 -5 get1den?6 -2 getwfk?6 -4 getwfq?6 -1 tolwfr?6 1.0d-8 # Underconverged : tolwfr 1.0d-22 should be used for converged results nbdbuf?6 2 # 2 buffer bands. RF converges much faster. #ieig2rf?6 1 ieig2rf?6 5 smdelta?6 1 rfphon?6 1 rfatpol?6 1 2 rfdir?6 1 1 1 iscf?6 -2 kptopt?6 -9 ndivsm?6 5 kptbounds?6 1/2 0.0 0.0 # L 0.0 0.0 0.0 # Gamma 0.0 1/2 1/2 # X 1/4 1/2 3/4 # W 3/8 3/8 3/4 # K 1/2 1/2 1/2 # L 1/4 1/2 3/4 # W 1/2 1/2 1.0 # X 3/8 3/8 3/4 # K 0.0 0.0 0.0 # Gamma # Cell dependent parameters acell 3*6.675 rprim 0 .5 .5 .5 0 .5 .5 .5 0 nsym 1 # Disable symmetries. Symmetries are not yet in production. natom 2 typat 1 1 xred 3*0.0 3*0.25 nband 12 ntypat 1 znucl 6 diemac 6 ecut 37 enunit 2 istwfk *1 pp_dirpath "$ABI_PSPDIR" pseudos "Pseudodojo_nc_sr_04_pw_standard_psp8/C.psp8" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% exclude_builders = [^s][^c][^o].* #%% [files] #%% files_to_test = #%% teph_tdep_legacy_4.abo, tolnlines= 1000, tolabs= 0.002, tolrel= 1.0e-03 #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = S. Ponc\'e #%% keywords = NC, DFPT, EPH_OLD #%% description = Temperature dependence calculation of diamond. #%%<END TEST_INFO>
Note the use of the usual input variables to define a path in the Brillouin Zone to build an electronic band structure: kptbounds, kptopt, and ndivsm. Note also that we have defined qptopt=3. The number of q-points is thus very easy to determine, as being the product of ngqpt values times nshiftq. Here a very rough 2*2*2 grid has been chosen, even less dense than the one for section 2.
Then issue:
abinit teph_tdep_legacy_4.abi
This is a significantly longer ABINIT run (5-10 minutes), also producing many files.
then use tests/tutorespfn/Input/teph_tdep_legacy_4_temperature.in for the python script.
1 2 temperature_4 0.1 0.1 0 0.5 0 1000 50 8 teph_tdep_legacy_4o_DS14_DDB teph_tdep_legacy_4o_DS24_DDB teph_tdep_legacy_4o_DS34_DDB teph_tdep_legacy_4o_DS44_DDB teph_tdep_legacy_4o_DS54_DDB teph_tdep_legacy_4o_DS64_DDB teph_tdep_legacy_4o_DS74_DDB teph_tdep_legacy_4o_DS84_DDB teph_tdep_legacy_4o_DS15_EIG.nc teph_tdep_legacy_4o_DS25_EIG.nc teph_tdep_legacy_4o_DS35_EIG.nc teph_tdep_legacy_4o_DS45_EIG.nc teph_tdep_legacy_4o_DS55_EIG.nc teph_tdep_legacy_4o_DS65_EIG.nc teph_tdep_legacy_4o_DS75_EIG.nc teph_tdep_legacy_4o_DS85_EIG.nc teph_tdep_legacy_4o_DS16_EIGR2D.nc teph_tdep_legacy_4o_DS26_EIGR2D.nc teph_tdep_legacy_4o_DS36_EIGR2D.nc teph_tdep_legacy_4o_DS46_EIGR2D.nc teph_tdep_legacy_4o_DS56_EIGR2D.nc teph_tdep_legacy_4o_DS66_EIGR2D.nc teph_tdep_legacy_4o_DS76_EIGR2D.nc teph_tdep_legacy_4o_DS86_EIGR2D.nc teph_tdep_legacy_4o_DS16_GKK.nc teph_tdep_legacy_4o_DS26_GKK.nc teph_tdep_legacy_4o_DS36_GKK.nc teph_tdep_legacy_4o_DS46_GKK.nc teph_tdep_legacy_4o_DS56_GKK.nc teph_tdep_legacy_4o_DS66_GKK.nc teph_tdep_legacy_4o_DS76_GKK.nc teph_tdep_legacy_4o_DS86_GKK.nc teph_tdep_legacy_4o_DS12_EIG.nc
with the usual syntax:
./temperature_final.py < teph_tdep_legacy_4_temperature.in
You can now copy the plotting script (Plot-EP-BS) python file from ~abinit/scripts/post_processing/plot_bs.py into the directory where you did all the calculations. Now run the script:
./plot_bs.py
with the following input data:
temperature_4_EP.nc
L \Gamma X W K L W X K \Gamma
-20 30
0
or more directly
./plot_bs.py < teph_tdep_legacy_4_plot_bs.abi
This should give the following bandstructure
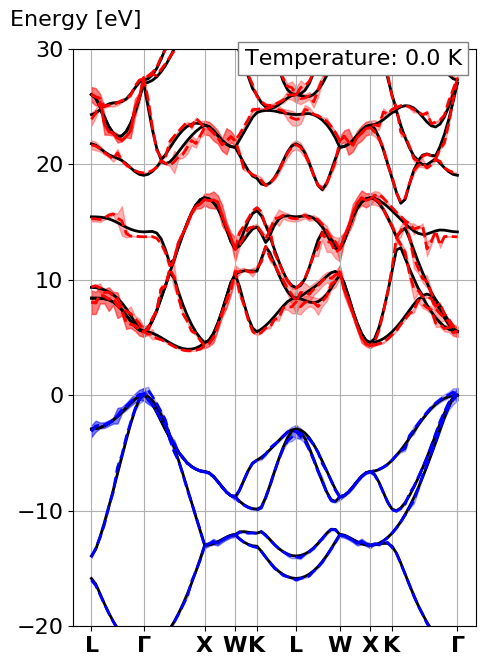
where the solid black lines are the traditional electronic bandstructure, the dashed lines are the electronic eigenenergies with electron-phonon renormalization at a defined temperature (here 0K). Finally the area around the dashed line is the lifetime of the electronic eigenstates.
Notice all the spikes in the electron-phonon case. This is because we did a completely under-converged calculation with respect to the q-point sampling.
It is possible to converge the calculations using ecut=30 Ha, a ngkpt grid of 6x6x6 and an increasing ngqpt grid to get converged results:
| Convergence study ZPR and inverse lifetime(1/τ) [eV] at 0K |
| q-grid | Nb qpt | Γ25' | Γ15 | Min Γ-X |
| | in IBZ | ZPR | 1/τ | ZPR | 1/τ | ZPR | 1/τ |
| 4x4x4 | 8 | 0.1175 | 0.0701 | -0.3178 | 0.1916 | -0.1570 | 0.0250 |
| 10x10x10 | 47 | 0.1390 | 0.0580 | -0.3288 | 0.1847 | -0.1605 | 0.0308 |
| 20x20x20 | 256 | 0.1446 | 0.0574 | -0.2691 | 0.1823 | -0.1592 | 0.0298 |
| 26x26x26 | 511 | 0.1448 | 0.0573 | -0.2736 | 0.1823 | -0.1592 | 0.0297 |
| 34x34x34 | 1059 | 0.1446 | 0.0573 | -0.2699 | 0.1821 | -0.1591 | 0.0297 |
| 43x43x43 | 2024 | 0.1447 | 0.0572 | -0.2650 | 0.1821 | -0.1592 | 0.0297 |
As you can see the limiting factor for the convergence study is the convergence of the LUMO band at \Gamma. This band is not the lowest in energy (the lowest is on the line between \Gamma and X) and therefore this band is rather unstable. This can also be seen by the fact that it has a large electronic broadening, meaning that this state will decay quickly into another state.
Using the relatively dense q-grid of 43x43x43 we can obtain the following converged bandstructure, at a high temperature (1900K):
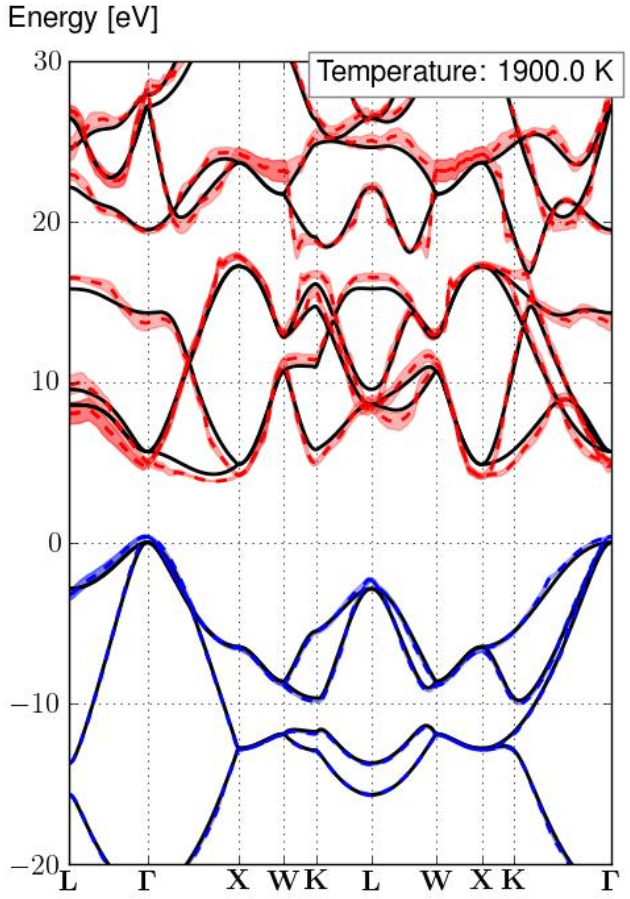
Here we show the renormalization at a very high temperature of 1900K in order to highlight more the broadening and renormalization that occurs. If you want accurate values of the ZPR at 0K you can look at the table above.
Important
If you use an extremely fine q-point grid, the acoustic phonon frequencies for q-points close to \Gamma will be wrongly determined by Abinit. Indeed in order to have correct phonon frequencies close to \Gamma, one has to impose the acousting sum rule with anaddb and asr@anaddb. However, this feature is not available in the python script. Instead, the script reject the contribution of the acoustic phonon close to \Gamma if their phonon frequency is lower than 1E-6 Ha. Otherwise one gets unphysically large contribution.
One can tune this parameter by editing the variable “tol6 = 1E-6” in the beginning of the script.
For example, for the last 43x43x43 calculation, it was set to 1E-4.
Important
It is possible to speed up the convergence with respect to increasing q-point density by noticing that the renormalization behaves analytically with increasing q-point grid and smaller broadening.
It is therefore possible to extrapolate the results. Different analytical behavior extists depending if the material is polar and if the state we are considering is a band extrema or not. More information can be found in Ref. [Ponce2015]